Voyage au cœur des argiles : quand la simulation révèle l'invisible
Publié par Sébastien Le Crom, le 25 juin 2026 350
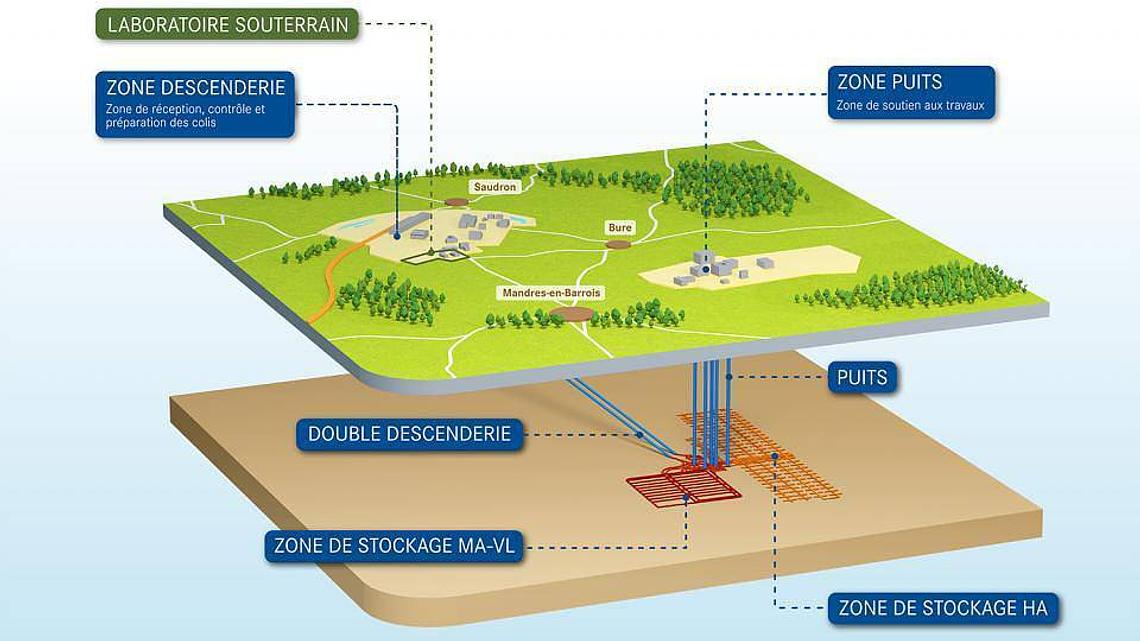
Image d'illustration : Andra (projet Cigéo) – https://www.andra.fr
L’argile comme prison pour les déchets radioactifs
Projet Cigéo : pourquoi enfouir les déchets radioactifs à 500 mètres sous terre ?
La France produit une grande partie de son électricité grâce à l’énergie nucléaire. Cette source d’énergie présente l’avantage d’être largement décarbonée, mais elle génère également des déchets radioactifs qu’il faut gérer sur le long terme afin de protéger les écosystèmes. Tous ces déchets ne posent toutefois pas les mêmes défis. Ils peuvent en effet présenter des niveaux de radioactivité très différents — mesurés par une grandeur appelée activité — ainsi que des durées de vie allant de quelques années à plusieurs centaines de milliers, voire plusieurs millions d’années ! Les solutions de gestion mises en œuvre diffèrent donc selon la nature des déchets considérés. Pour les déchets les plus problématiques, c’est-à-dire ceux de haute activité et à vie longue, la solution retenue en France est le stockage géologique profond dans le cadre du projet Cigéo, développé par l’Andra (Agence nationale pour la gestion des déchets radioactifs). [1]
Les déchets sont d’abord confinés dans des colis spécialement conçus pour résister à leur environnement. Ces colis comportent notamment des matériaux métalliques, en particulier des aciers, qui contribuent à la rétention des substances radioactives. Toutefois, aucun matériau artificiel ne peut garantir un confinement parfait sur des dizaines ou des centaines de milliers d’années. Le stockage géologique repose donc sur le principe que plusieurs barrières successives contribuent à limiter et ralentir une éventuelle migration des radioéléments : on parle de stratégie de barrières multiples. Les colis sont ainsi placés dans des galeries souterraines situées à environ 500 mètres de profondeur, au sein d’une formation géologique naturelle constituée d’une épaisse couche d’argile. Cette dernière constitue la barrière ultime du système de stockage et la dernière ligne de défense contre une éventuelle migration des radioéléments dans l’environnement.
Ce choix n’est pas dû au hasard. Les argiles possèdent plusieurs propriétés particulièrement intéressantes pour le stockage géologique, dont leur très faible perméabilité. Dans ces matériaux, le déplacement de l’eau et des espèces chimiques dissoutes s’effectue principalement par diffusion qui, contrairement à un transfert par écoulement, est un mécanisme naturellement lent, pouvant nécessiter des temps extrêmement longs pour parcourir de grandes distances.
Quand les déchets radioactifs produisent du gaz
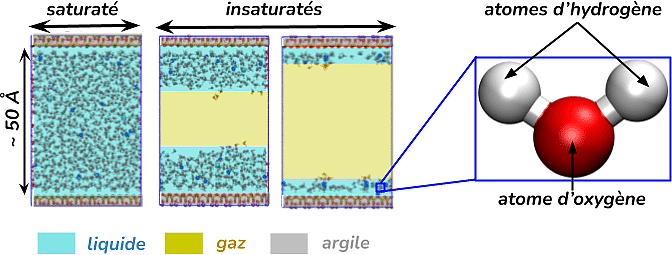
Cependant, l’environnement de stockage n’est pas figé. Au cours du temps, différents phénomènes comme la corrosion des matériaux métalliques ou la radiolyse de l’eau sous l’effet des rayonnements vont à la fois dégrader les matériaux des colis et produire des gaz, en particulier du dihydrogène (H₂). Une partie de ce gaz peut alors pénétrer dans l’argile au voisinage des galeries et modifier la façon dont l’eau se répartit à l’intérieur du matériau. Le milieu, initialement rempli d’eau et d’ions, devient progressivement partiellement occupé par du gaz, créant un système diphasique liquide-gaz : on parle alors de conditions insaturées.
Cette évolution soulève une question importante : comment la présence de gaz influence-t-elle le transport de l’eau et des espèces dissoutes dans les argiles ? La réponse à cette question est essentielle afin d’améliorer la prédiction à long terme des transferts au sein des formations géologiques utilisées pour le stockage des déchets radioactifs.
Les argiles : une barrière naturelle multi-échelles
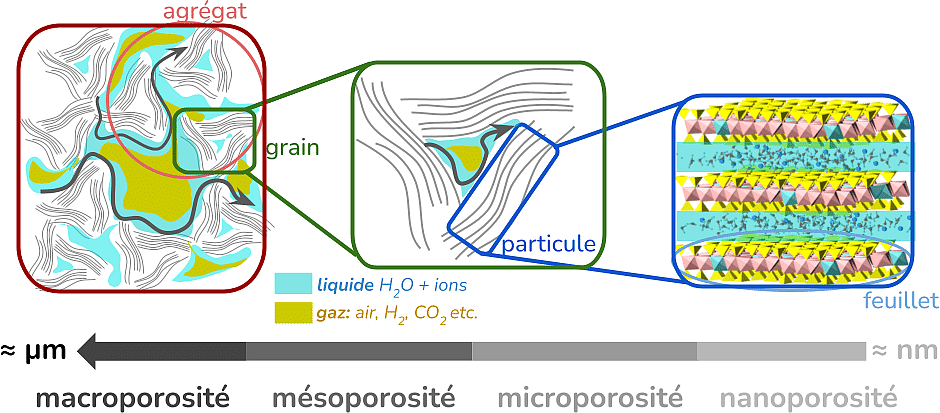
Les argiles possèdent une structure hiérarchique particulièrement complexe, organisée sur plusieurs échelles de taille. Elles sont constituées de feuillets minéraux extrêmement fins — quelques nanomètres d’épaisseur — qui s’assemblent pour former des particules, elles-mêmes regroupées en agrégats. Entre ces différentes structures existent des espaces vides appelés pores. Leur taille varie de quelques nanomètres à plusieurs micromètres et ils constituent les voies de circulation de l’eau, des ions et éventuellement du gaz.
À l’échelle microscopique, les molécules d’eau et les ions interagissent en permanence avec les surfaces minérales. Ces surfaces argileuses sont chargées négativement et agissent donc un peu comme des aimants, attirant les ions de charge positive tout en repoussant ceux de charge négative. Les interactions entre l’eau, les ions et les surfaces minérales déterminent alors la façon dont la phase aqueuse s’organise dans les pores, les chemins de diffusion que les espèces dissoutes peuvent emprunter et, au final, la vitesse à laquelle elles se déplacent dans l’environnement. La présence de gaz dans les pores modifie cet équilibre et perturbe les chemins de diffusion disponibles. Les conséquences peuvent être très différentes selon les espèces considérées, notamment en fonction de leur charge électrique.
Les
phénomènes de transport que l’on cherche à prédire se déroulent
à des échelles gigantesques comparées à celles des atomes :
plusieurs kilomètres et parfois plusieurs millions
d’années. Pourtant, les mécanismes qui les contrôlent
trouvent leur origine à des échelles infiniment plus petites. Pour
comprendre ces mécanismes fondamentaux, il faut donc être capable
d’observer ce qui se passe à l’échelle des atomes et des
molécules. C’est précisément ce que permet la dynamique
moléculaire, une technique de simulation numérique
qui peut être utilisée comme un véritable microscope virtuel
capable de suivre individuellement le mouvement de chaque atome d’un
système.
La simulation moléculaire : plus puissant que le plus puissant microscope !
Observer directement ce qui se passe à l’intérieur des pores d’une argile est un véritable défi. Même les microscopes les plus performants ne disposent pas d’une résolution suffisante pour suivre individuellement le mouvement des molécules d’eau et des ions dans ces espaces extrêmement confinés. Pour contourner cette difficulté, il est possible de recourir à la modélisation moléculaire, dont la dynamique moléculaire constitue l’une des approches les plus utilisées. Grâce à cette technique de simulation numérique, il devient possible de suivre le mouvement de chaque atome d’un système.
Le principe est relativement simple puisqu’il repose sur la mécanique classique de Newton et en particulier sur le principe fondamental de la dynamique (PFD), enseigné dès le lycée. Le PFD permet de calculer le mouvement d’un objet à partir de sa masse et des forces qui s’exercent sur lui. Dans une simulation de dynamique moléculaire, les « objets » auxquels sont appliquées les équations de Newton sont les atomes. Chaque atome est représenté par une sphère possédant une masse, une taille et une charge électrique permettant de reproduire le comportement de l’élément chimique considéré. Une molécule d’eau, par exemple, est modélisée par trois sphères représentant les trois atomes qui la constituent : un atome d’oxygène et deux atomes d’hydrogène. Dans les systèmes étudiés dans ce projet, plusieurs milliers à plusieurs dizaines de milliers d’atomes représentant le feuillet d’argile, l’eau, les ions et parfois le gaz sont ainsi simulés simultanément.
Au cours de la simulation, ces atomes interagissent continuellement les uns avec les autres. Certaines interactions les attirent, d’autres les repoussent, ce qui les fait se déplacer et se réorganiser en permanence. En répétant ces calculs des milliards de fois, il devient possible de reconstruire l’évolution complète du système au cours du temps et d’observer des phénomènes qui seraient autrement invisibles.
Les simulations produisent alors d’immenses quantités de données contenant la position de chaque atome à chaque instant. L’ensemble de ces positions successives constitue ce que l’on appelle une trajectoire. À partir de ces trajectoires, il est possible de calculer des propriétés directement liées au comportement du matériau : organisation de l’eau dans les pores, répartition des ions ou encore mobilité des espèces dissoutes. La mobilité des espèces peut notamment être quantifiée grâce aux coefficients de diffusion, qui mesurent la vitesse à laquelle les espèces se déplacent dans le matériau. En s’appuyant sur des outils issus de la physique statistique, la dynamique moléculaire permet ainsi de relier les interactions entre les atomes à l’échelle microscopique aux propriétés macroscopiques observées à l’échelle du matériau.
Dans le cadre de ce projet, cette approche a été utilisée pour comprendre comment la présence de gaz dans les pores de l’argile modifie l’organisation de l’eau et des ions ainsi que le transport des espèces dissoutes.
Quand le gaz bouleverse l’organisation des ions
Dans un pore entièrement saturé en eau, c’est-à-dire ne contenant qu’une phase liquide composée d’eau et d’ions, les surfaces argileuses chargées négativement attirent naturellement les ions de charge positive présents dans la solution. Ces ions, appelés cations, s’accumulent ainsi au voisinage des surfaces afin de compenser la charge négative de l’argile. Cette organisation particulière des charges électriques à proximité de l’argile est connue sous le nom de double couche électrique. Plus on s’éloigne de la surface, plus son influence diminue. Ainsi, au centre du pore, l’eau et les ions retrouvent un comportement proche de celui observé dans un liquide non perturbé par la présence d’une surface : ils ne « voient » plus l’argile.
Lorsque du gaz apparaît dans le système, sa répartition n’est pas aléatoire. Les simulations montrent que la phase gazeuse se localise systématiquement au centre du pore tandis que l’eau et les ions restent adsorbés au voisinage des surfaces argileuses. Cette organisation résulte des interactions attractives entre les cations et les surfaces argileuses, ainsi que de l’affinité des ions pour l’eau qui les entoure. En effet, comme on l’a vu précédemment, les ions positifs sont attirés par la surface chargée négativement. De plus, ils ne peuvent pas passer directement dans la phase gazeuse : ils doivent rester hydratés, c’est-à-dire entourés de molécules d’eau. Les cations restent donc à proximité des surfaces tout en conservant leur sphère d’hydratation. Les molécules d’eau demeurent ainsi majoritairement localisées au voisinage de l’argile. Le gaz occupe alors préférentiellement le centre du pore, là où l’influence de l’argile est la plus faible.
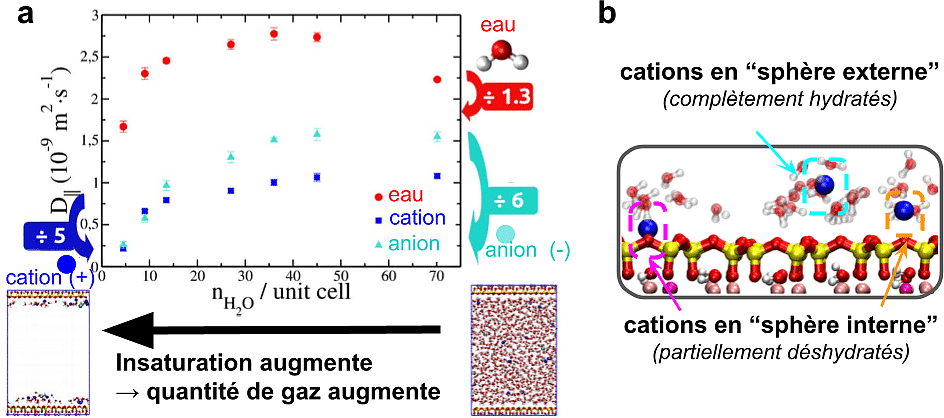
À mesure que le taux de saturation diminue, la quantité de gaz présente dans le pore augmente et le volume de la phase aqueuse se réduit progressivement. Les régions situées au centre du pore, les moins influencées par les surfaces argileuses, disparaissent alors en premier et les ions sont progressivement contraints de se rapprocher de l’argile. Cette compression de la phase aqueuse perturbe profondément l’organisation initiale de la double couche électrique.
Les cations étant déjà majoritairement localisés à proximité de l’argile dans l’état saturé, ils sont relativement peu affectés dans un premier temps par cette compression de la phase aqueuse. La situation est différente pour les anions. Initialement localisés plus loin des surfaces en raison de la répulsion électrostatique exercée par l’argile, ils sont particulièrement affectés par la diminution de l’espace disponible. En se rapprochant de l’argile, ils subissent des interactions répulsives de plus en plus fortes avec la charge négative du minéral. Afin de réduire ces répulsions, les cations positifs s’intercalent entre les anions et la surface, jouant en quelque sorte le rôle d’écran électrostatique.
Lorsque le taux de saturation devient très faible, l’espace disponible devient insuffisant pour maintenir tous les ions totalement hydratés. Une partie des cations perd alors une fraction des molécules d’eau qui les entourent et vient interagir directement avec les atomes de surface de l’argile. On parle alors de cations de sphère interne, car aucune couche d’eau ne les sépare plus de la surface du minéral.
L’insaturation ne modifie donc pas uniquement la quantité d’eau présente dans les pores. Elle réorganise profondément la distribution des ions et la manière dont la charge négative des surfaces argileuses est compensée. Cette nouvelle organisation des charges influence directement la mobilité des espèces dissoutes et, par conséquent, leur transport dans les argiles.
Du réarrangement des ions au ralentissement de la diffusion
En dynamique moléculaire, la mobilité des espèces est généralement caractérisée à l’aide des coefficients de diffusion, qui mesurent la vitesse moyenne à laquelle molécules et ions se déplacent dans le matériau. Plus le coefficient de diffusion est élevé, plus l’espèce se déplace rapidement.
Les simulations montrent que la diminution du taux de saturation s’accompagne d’un ralentissement global du transport de l’eau et des ions. Cependant, toutes les espèces ne sont pas affectées de la même manière et l’ampleur du ralentissement dépend fortement de leur charge. Ce sont les ions qui présentent les diminutions de mobilité les plus importantes. Lorsque la quantité de gaz augmente, l’espace disponible pour la phase aqueuse se réduit progressivement. Les ions, qui ne peuvent exister que dans l’eau, se retrouvent alors confinés dans un volume de plus en plus petit. Ils sont contraints de se rapprocher les uns des autres ainsi que des surfaces argileuses. Les interactions électrostatiques deviennent alors plus importantes, ce qui limite progressivement leur mobilité.
Les anions sont particulièrement sensibles à l’insaturation. Initialement localisés dans la région centrale du pore, ils voient leur espace de diffusion fortement réduit à mesure que cette région est envahie par le gaz ce qui entraîne une forte diminution de leur mobilité. En effet, les anions sont progressivement comprimés entre la phase gazeuse, qui occupe une fraction croissante du pore, et les surfaces argileuses chargées négativement dont ils sont repoussés. L’espace réellement accessible à leur diffusion devient alors de plus en plus limité.
Les cations sont eux aussi ralentis, mais pour des raisons légèrement différentes. Aux plus faibles saturations, une fraction croissante des cations se retrouve sous forme de cations de sphère interne, directement liés à la surface du minéral. Cette évolution contribue également à réduire leur mobilité globale. En effet, ces ions sont beaucoup moins libres de se déplacer que les cations restant totalement hydratés dans la phase aqueuse.
Le comportement de l’eau est plus subtil. Contrairement aux ions, les molécules d’eau ne sont pas contraintes de rester exclusivement dans la phase liquide. Elles peuvent également se trouver à l’interface entre l’eau et le gaz et échanger continuellement entre ces deux environnements. Certaines molécules d’eau peuvent occasionnellement explorer la région gazeuse du pore, où elles sont beaucoup moins contraintes que dans la phase liquide. Cette situation favorise leur mobilité et peut même conduire à une légère augmentation du coefficient de diffusion pour certains états de saturation intermédiaires. Lorsque la quantité d’eau devient très faible, l’effet du confinement finit néanmoins par dominer et la diffusion de l’eau diminue à son tour.
Ces résultats montrent que l’apparition du gaz ne se contente pas de réduire la quantité d’eau présente dans les pores. En comprimant progressivement la phase aqueuse, elle réorganise profondément la distribution des ions et modifie leur espace de diffusion. Cette réorganisation se traduit par un ralentissement important de la mobilité des espèces dissoutes, particulièrement marqué pour les ions. À l’échelle moléculaire, l’insaturation modifie donc profondément les mécanismes de transport dans les argiles. Ces effets doivent être pris en compte pour améliorer les modèles utilisés à plus grande échelle dans les études de sûreté du stockage géologique.
De la molécule au stockage géologique
Les simulations de dynamique moléculaire permettent d’accéder à des informations extrêmement détaillées sur le comportement de l’eau et des ions à l’échelle atomique. Elles permettent par exemple de comprendre comment la présence de gaz modifie l’organisation de la phase aqueuse, la répartition des ions ou encore leur mobilité. Cependant, ces simulations restent limitées à des systèmes de quelques nanomètres à quelques dizaines de nanomètres et à des temps allant de la nanoseconde à la microseconde.
Or, lorsqu’il s’agit d’évaluer la sûreté d’un stockage géologique comme Cigéo, les échelles considérées sont tout autres : plusieurs centaines de mètres à plusieurs kilomètres et des temps pouvant atteindre plusieurs millions d’années. Il est donc impossible d'utiliser directement la dynamique moléculaire pour prédire le comportement du système à ces échelles. En effet, l’approche présentée jusque-là permet de décrire avec précision le comportement de l’eau et des ions à l’intérieur d’un pore de taille donnée. Cependant, comme on l’a vu précédemment, les argiles réelles sont constituées d’un vaste réseau de pores de tailles variées, connectés les uns aux autres. Les espèces dissoutes ne diffusent donc pas dans un unique pore isolé mais empruntent des chemins complexes à travers l’ensemble du matériau.
Ces chemins de diffusion dépendent fortement de la structure du réseau poreux mais également de la charge des espèces considérées. Par exemple, les ions négatifs sont partiellement exclus des pores les plus étroits en raison de la charge négative des surfaces argileuses, tandis que les cations peuvent y pénétrer plus facilement. Deux espèces différentes peuvent ainsi emprunter des chemins de diffusion très différents au sein d’un même matériau, ce qui a une conséquence directe sur la rapidité de leur diffusion dans le milieu argileux.
Représenter explicitement un tel réseau poreux nécessiterait de suivre simultanément plusieurs millions, voire plusieurs milliards d’atomes, ce qui reste aujourd’hui hors de portée des simulations de dynamique moléculaire. Il devient donc nécessaire de développer des modèles capables d’atteindre des échelles spatiales et temporelles beaucoup plus grandes tout en conservant les informations essentielles obtenues à l’échelle moléculaire. Cette démarche est appelée l’« up-scaling ».
Les informations obtenues par dynamique moléculaire peuvent alors être utilisées pour alimenter des modèles à plus grande échelle. Les propriétés calculées à l’échelle microscopique — répartition du gaz et de l’eau, organisation des ions, structure de la double couche électrique ou encore coefficients de diffusion — servent alors de données d'entrée pour des simulations capables de traiter des systèmes beaucoup plus grands.
De l’échelle mésoscopique...

Une première étape consiste à réaliser des simulations à l'échelle mésoscopique, typiquement sur des systèmes de quelques micromètres et sur des temps pouvant atteindre la milliseconde voire davantage. À cette échelle, il devient possible de représenter explicitement un assemblage de particules d'argile et une distribution réaliste des pores.
Les espèces dissoutes ne sont alors plus décrites atome par atome. Leur déplacement est simulé par dynamique brownienne, une méthode qui reproduit statistiquement les mouvements calculés à l’échelle moléculaire tout en permettant d’atteindre des systèmes beaucoup plus grands. L’objectif est de suivre le transport de l’eau et des ions dans un réseau poreux beaucoup plus réaliste et d’intégrer des effets absents des simulations moléculaires, comme la connectivité entre les pores ou la tortuosité des chemins de diffusion à l’échelle du matériau.
Ces simulations permettent de calculer directement des coefficients de diffusion comparables aux mesures expérimentales. À l'heure actuelle, cette approche reproduit correctement les expériences réalisées en conditions saturées. En revanche, elle surestime encore fortement la diffusion observée dans les systèmes partiellement saturés. Comprendre l'origine de cet écart constitue aujourd'hui un verrou scientifique important.
Une thèse débutant à l'automne 2026 visera précisément à mieux décrire, via des simulations de dynamique moléculaire, la répartition de l'eau et du gaz dans les pores insaturés et à intégrer ces nouvelles connaissances dans les modèles mésoscopiques. Les travaux porteront notamment sur l'effet de la géométrie des pores, de la structure des surfaces argileuses et de leur charge électrique sur l'organisation de l'eau et la diffusion des espèces dissoutes.
Jusqu'aux modèles macroscopiques !
Même l'échelle mésoscopique reste encore très éloignée des dimensions et des durées associées au stockage géologique. Pour prédire le devenir des radionucléides sur plusieurs kilomètres et plusieurs centaines de milliers d'années, les spécialistes des milieux géologiques utilisent d’autres types de modèles que l’on appelle modèles de transport réactif. [4]
Ces modèles décrivent simultanément le transport des espèces chimiques et les réactions chimiques qu'elles peuvent subir au cours de leur déplacement. Ils constituent aujourd'hui les principaux outils utilisés pour évaluer à long terme les performances des barrières géologiques dans le cadre du stockage des déchets radioactifs.
À chaque changement d'échelle, une partie de la description détaillée du système est remplacée par une représentation plus simple. La dynamique moléculaire calcule explicitement les interactions entre les atomes. Les modèles mésoscopiques utilisent ensuite les propriétés moyennes obtenues à cette échelle pour décrire le déplacement des espèces dans un réseau poreux complexe. Enfin, les modèles de transport réactif ne suivent plus individuellement les molécules mais utilisent des propriétés effectives, comme les coefficients de diffusion, pour prédire le comportement du système à l'échelle du stockage.
C'est précisément cette chaîne de modèles, allant de l'atome jusqu'au stockage géologique, qui permet aujourd'hui de relier les mécanismes fondamentaux observés à l'échelle moléculaire aux questions de sûreté posées à l'échelle de Cigéo.
Alors, à quoi servent encore les expériences ?
À ce stade, une question légitime peut se poser : si la simulation numérique permet d'observer chaque atome individuellement, d'accéder à des informations impossibles à mesurer expérimentalement et même de prédire le comportement d'un système sur des milliers voire des millions d'années, pourquoi continue-t-on à faire des expériences ?
La réponse est simple : une simulation n'est pas la réalité !
Quel que soit le niveau de description considéré — dynamique moléculaire, dynamique brownienne ou transport réactif — les simulations reposent toujours sur des modèles. Or, aussi sophistiqués soient-ils, ces modèles ne sont que des représentations simplifiées du monde réel. Ils tentent de reproduire la réalité le plus fidèlement possible, mais ils restent nécessairement imparfaits.
La force de la modélisation est justement qu'elle permet d'accéder à des informations inaccessibles expérimentalement. En dynamique moléculaire, il est possible de suivre individuellement le mouvement de chaque atome et d'analyser les interactions qui contrôlent le comportement du système. À l'autre extrémité des échelles, les modèles de transport réactif permettent d'explorer des temps de plusieurs centaines de milliers d'années, voire davantage, ce qu'aucune expérience ne pourrait reproduire directement.
Mais ces capacités extraordinaires reposent toujours sur des hypothèses et des approximations. En dynamique moléculaire, le modèles central est le champ de force qui décrit les interactions entre les atomes. C’est lui qui détermine les forces appliquées aux atomes et donc l'ensemble de leur trajectoire au cours de la simulation. Si ces interactions sont mal décrites, les mouvements calculés seront eux aussi incorrects, et les propriétés obtenues à partir des simulations s'éloigneront progressivement du comportement réel du système.
Il n'existe d'ailleurs pas un unique champ de force pour décrire un matériau donné. Plusieurs modèles différents peuvent être utilisés pour simuler un même système. Certains privilégient la rapidité de calcul afin de pouvoir étudier des systèmes plus grands ou des temps plus longs. D'autres cherchent au contraire à représenter les interactions de manière plus réaliste au prix d'un coût numérique plus important. Chaque approche possède ses avantages, ses limites et son domaine de validité.
Une question que nous n'avons volontairement pas abordée ici est celle de l'influence du choix du champ de force sur les résultats obtenus. Deux modèles différents peuvent parfois conduire à des prédictions sensiblement différentes pour un même système. L'étude de cette question dans le contexte des argiles partiellement saturées constitue un sujet de recherche à part entière qui pourrait faire l'objet d'un futur article sur Échosciences Pays de la Loire.
Finalement, l'objectif de la modélisation n'est pas de remplacer l'expérience, mais de travailler avec elle. L'expérience permet de confronter les modèles à la réalité, tandis que la simulation permet d'explorer ce que l'expérience ne peut pas observer directement. C'est de ce dialogue permanent entre expériences et simulations que naissent les progrès scientifiques et notre capacité à prédire le comportement de systèmes aussi complexes que les argiles utilisées pour le stockage géologique des déchets radioactifs.
Références
[1] ANDRA. Les recherches de l’Andra sur le stockage géologique des déchets radioactifs à haute activité et à vie longue. Dossier 2005, 2005.
[2] Le Crom, S.; Tournassat, C.; Robinet, J.-C. & Marry, V. Influence of Water Saturation Level on Electrical Double Layer Properties in a Clay Mineral Mesopore: A Molecular Dynamics Study. The Journal of Physical Chemistry C, 126, 1, 647–654, 2022.
[3] Ferrage, E.; Hubert, F.; Dabat, T. et al. Anisotropy in Particle Orientation Controls Water Diffusion in Clay-Rich Porous Media. Applied Clay Science, 244, 107117, 2023.
[4] Tournassat, C. & Steefel, C. I. Ionic Transport in Nano-Porous Clays with Consideration of Electrostatic Effects. Reviews in Mineralogy and Geochemistry, 80, 1, 287–329, 2015.